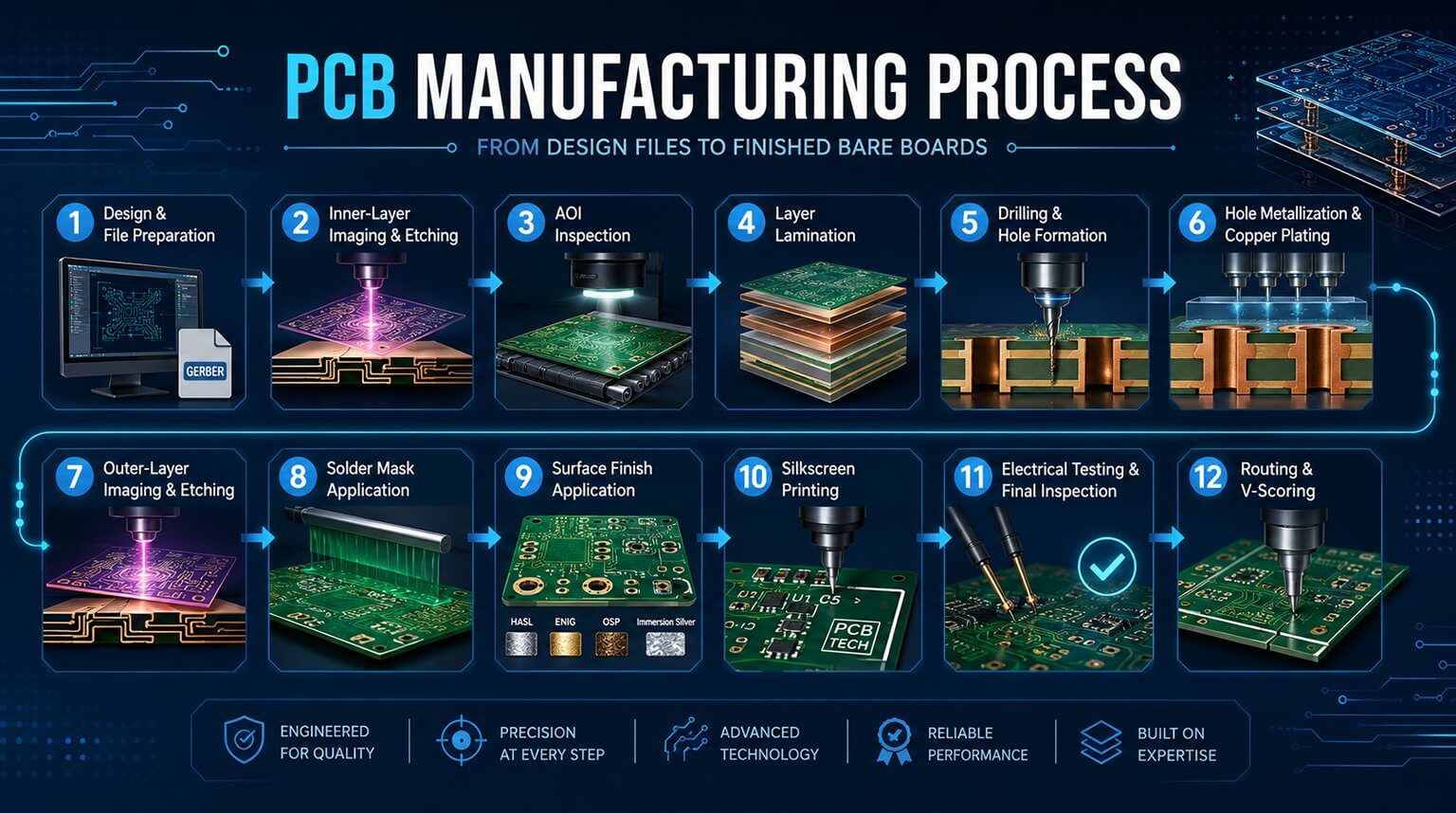
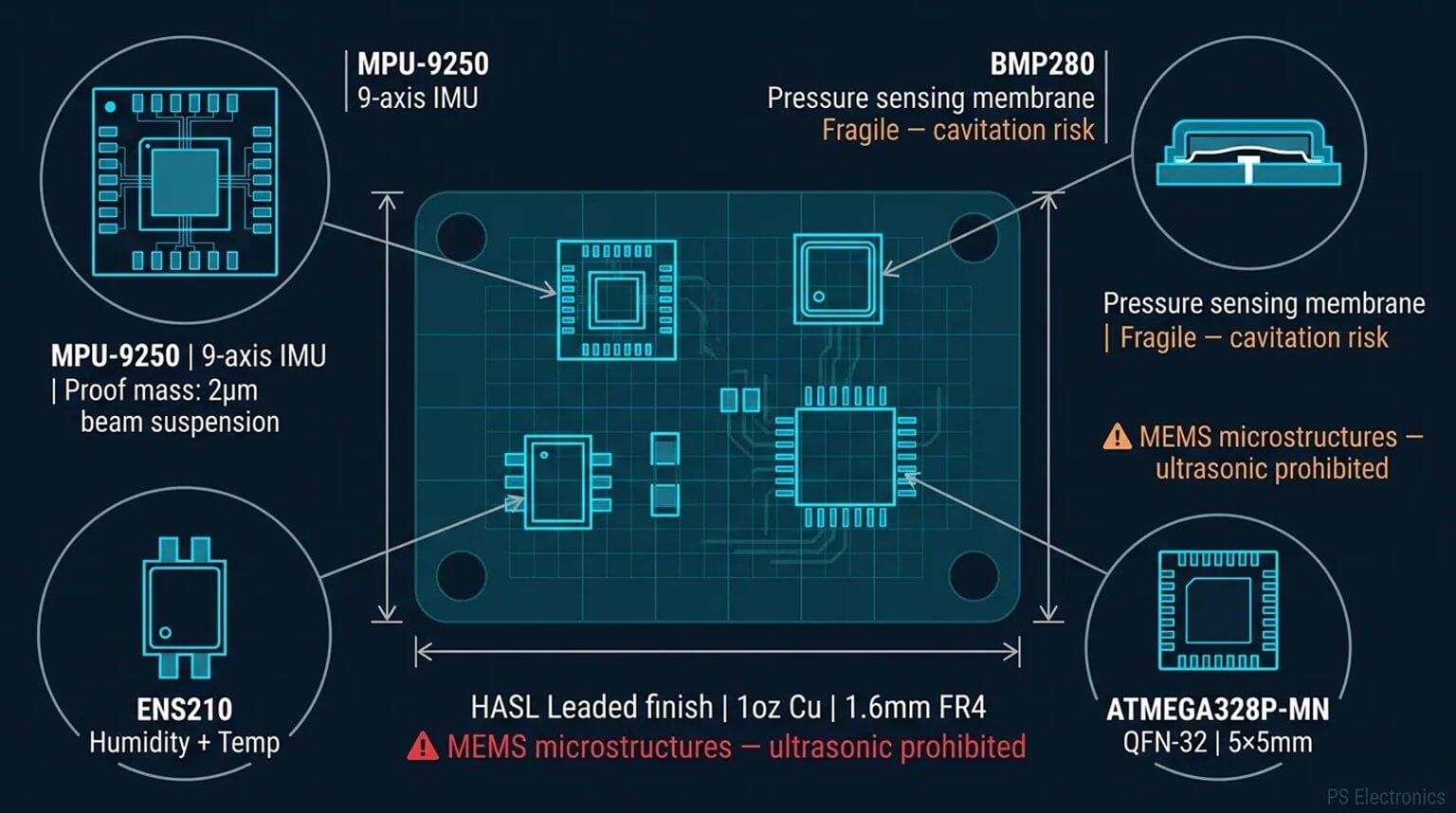
In the medical device industry, quality and safety are non-negotiable priorities. ISO 13485, a dedicated quality management system (QMS) standard for this sector, serves as the core guideline for compliance, market access, and risk management worldwide. Whether it’s active devices, passive implants, in vitro diagnostic reagents, or medical software, ISO 13485 spans the entire product lifecycle—from design and development, through manufacturing, to post-market traceability. It defines the “universal language” of quality control within the medical device industry.
This article by PCBCool will thoroughly deconstruct ISO 13485 from several dimensions: the origin of the standard, its evolution, core requirements, scope of application, and certification value. This will help professionals gain a deeper understanding of its core logic and key implementation points.
ISO 13485, officially titled Medical devices—Quality management systems—Requirements for regulatory purposes, is a specialized standard developed by the International Organization for Standardization (ISO) under its technical committee for medical devices (ISO/TC 210). The primary focus of ISO 13485 is “regulatory compliance at the core and risk management as the tool,” providing a practical framework for quality management across the entire medical device industry chain to ensure that products meet global regulatory requirements and clinical safety and efficacy demands.
Unlike general quality management standards such as ISO 9001, ISO 13485 downplays the universal requirement for “continuous improvement” and places a stronger emphasis on the mandatory “maintenance of system effectiveness” and “regulatory conformity.” It is not a standalone quality norm but needs to deeply integrate the various national regulatory frameworks for medical devices (such as China’s NMPA regulations, the EU’s MDR, and the US FDA QSR 820). The core goal is to standardize management processes to reduce safety risks throughout the product lifecycle, ensuring the health and safety of patients and users.
It is important to note that ISO 13485 is an independent standard system. Although it draws from the PDCA (Plan-Do-Check-Act) cycle concept of ISO 9001, it introduces numerous specific requirements tailored to the unique needs of the medical device industry, covering critical areas such as risk management, sterilization control, UDI traceability, and adverse event handling. This makes it an essential “passport” for medical device companies seeking to enter global markets.
The development of ISO 13485 has always been synchronized with global regulatory upgrades in the medical device sector and technological innovations. The standard has undergone three major revisions, progressively expanding its coverage of the entire industry chain and new technologies, resulting in the current version of the standard:
In 1996, ISO released the first edition of ISO 13485, replacing the relevant content from the ISO 8402 standard concerning medical devices. The core value of this edition was to “establish a specialized framework for the industry.” It introduced specific terminology and basic requirements for medical device quality management systems, aligning with the regulatory frameworks in place at the time in Europe and North America. This edition provided a basis for the industry to move beyond the limitations of general quality standards. However, the terms were relatively basic, and the focus was primarily on quality control during the manufacturing stage, without covering the full lifecycle or risk management.
In 2003, ISO conducted the first significant revision, releasing ISO 13485:2003. The key changes in this version were the “strengthening of regulatory compatibility and risk management fundamentals.” This edition explicitly defined the core purpose of “regulatory requirements” and integrated the foundations of risk management. It also detailed specific requirements for design and development, manufacturing, and testing, aligning more closely with the EU MDD (Medical Device Directive) and the US FDA QSR 820 at the time.
In March 2016, ISO published ISO 13485:2016, which came into effect in 2017. The core upgrades in this edition were focused on “lifecycle management and global regulatory harmonization.” This version expanded the penetration of risk management throughout the entire process, mandating the integration of ISO 14971 (the medical device risk management standard). It also added specific requirements for supply chain management, product traceability, and software control. This version aligned more closely with global regulatory trends and removed redundant clauses that overlapped with ISO 9001.
On May 9, 2025, ISO will officially release ISO 13485:2025, marking the entry of medical device quality management into a new era of “digitalization, intelligence, and full-chain compliance.” This revision is a comprehensive update to the 2016 edition and focuses on four key areas:
- The addition of clauses on ethics and data security for AI-based medical devices, adapting to new technologies such as Software as a Medical Device (SaMD) and 3D-printed medical devices.
- Strengthened electronic record control, including the use of blockchain and other technologies to prevent data tampering, in compliance with FDA 21 CFR Part 11 and EU Annex 11.
- Upgraded supply chain management, implementing tiered audits and mandatory on-site inspections, extending control to raw material suppliers.
- Extending traceability to 10 years and enhancing post-market surveillance (PMS) mechanisms.
ISO 13485’s clause structure focuses on “regulatory compliance, risk control, and traceability,” covering 8 major chapters. It can be broken down into five core management modules and four special control requirements, balancing general applicability with industry-specific needs. The 2025 version further strengthens digitalization and adaptability to new technologies, becoming a key focus for audits.
The core requirement is for organizations to establish a clear quality management framework, define roles and responsibilities, and ensure that top management is committed to regulatory compliance and resource allocation.
The 2025 version adds a new requirement: the CEO must chair quarterly reviews of regulatory compliance, including evaluations of AI ethics compliance. It also requires companies to establish quality policies and goals that are measurable, actionable, and directly linked to product safety risks and regulatory requirements. Furthermore, a management review mechanism must be established to evaluate the system’s effectiveness regularly, addressing compliance risks and quality issues promptly.
This module focuses on the five main resources: people, equipment, materials, methods, and environment.
For personnel, key positions (such as sterilization inspection, risk assessment, and product review) must be staffed by individuals with qualifications recognized by NMPA (China’s National Medical Products Administration), and regular training on medical device regulations and quality control skills must be conducted, with complete records maintained.
Regarding infrastructure, production and inspection equipment must be calibrated and maintained regularly to ensure accuracy. Specialized equipment (such as 3D printing and AI detection tools) must have a digital verification process in place. For the work environment, sterile product manufacturing must be carried out in certified cleanrooms, with the 2025 version requiring real-time monitoring of temperature and humidity and cloud-based data uploading for traceability. Additionally, a resource assurance mechanism should be in place to ensure that necessary funds, technology, and personnel are available for quality control, avoiding quality risks due to insufficient resources.
This is the core module of ISO 13485, covering the entire process from design and development to purchasing, manufacturing, and after-sales services, with specific compliance requirements for each stage:
- Design and Development: A comprehensive design control process must be established, specifying design inputs (including patient safety targets), outputs, reviews, and validation/verification requirements. The 2025 version adds requirements for AI algorithm validation and 3D printing parameter records. Design control documentation (DHF) should include data on biocompatibility testing and algorithm validation to ensure designs meet regulatory and safety standards.
- Purchasing: Suppliers must be evaluated and controlled through tiered assessments, with high-risk suppliers undergoing on-site audits. Suppliers must demonstrate UDI (Unique Device Identification) traceability and sign quality agreements that define material quality standards and traceability requirements. Implants must trace materials back to the raw material level.
- Production: Standard operating procedures (SOPs) must be developed to control process parameters. “Single-unit traceability” (complete UDI coverage) must be implemented to eliminate deviations during production. For sterile products, sterilization processes must be strictly controlled, with complete sterilization records retained.
- Services: A post-market service process must be established to handle customer complaints and quality issues promptly. Feedback must be collected for post-market surveillance, with the 2025 version requiring service records to be linked to the product traceability system for full lifecycle traceability.
Companies must establish monitoring and analysis mechanisms to continually track the effectiveness of the quality management system, product quality, and regulatory compliance:
- Inspections must be conducted on every batch of products, with inspection records kept for at least five years.
- A system for collecting and reporting adverse events must be in place, with adverse events reported within 24 hours to regulatory platforms.
- Data analysis should be used to identify quality and compliance risks, with analytical reports forming the basis for corrective actions.
- A corrective and preventive action (CAPA) system must be in place to address identified issues, with clear responsibility, timelines, and verification requirements. The 2025 version requires the analysis of adverse event trends to be included in management reviews to enhance risk warning capabilities.
Companies must establish a product recall management system, defining recall processes, responsibilities, and emergency plans. A simulated recall exercise should be conducted at least once per year, and recall rates should be included in annual management reviews. Additionally, continuous quality system improvements should be based on management reviews, data analysis, and customer feedback, ensuring a “compliance-optimization-re-compliance” cycle. Unlike ISO 9001, the focus here is more on maintaining regulatory compliance rather than purely performance enhancement. The core goal is to ensure that the quality management system remains up-to-date with the latest regulatory requirements, minimizing compliance risks.
Since the 2016 version, ISO 13485 has mandated that companies integrate ISO 14971 (the medical device risk management standard) into their quality management systems. The 2025 version further refines the requirements for risk control. The fundamental logic is “lifecycle risk management,” which covers risk identification, assessment, and control from design through to production and post-market monitoring. Risk control measures must be documented, verifiable, and traceable.
For AI-based devices, the 2025 version introduces the requirement for “algorithm risk classification,” where high-risk algorithms must undergo third-party verification. For implantable devices, long-term risk monitoring for up to 10 years is required, with the risk data maintained.
For sterile medical devices (such as syringes, joint implants, sterile dressings), ISO 13485 sets out specific control requirements. The 2025 version improves control over clean environments and sterilization processes:
- Cleanrooms must meet ISO 14644 standards, and cleanliness levels must be defined according to product risk classifications. Real-time monitoring of suspended particles and microorganisms is required, with data uploaded to regulatory platforms.
- Sterilization processes must use validated methods (such as steam or irradiation), with sterilization parameters and validation reports retained to ensure repeatability and traceability of the sterilization process.
- A sterile barrier system must be in place to prevent contamination during storage and transportation, and packaging materials for sterile products must undergo biocompatibility testing.
Unique Device Identification (UDI) is central to the ISO 13485 traceability system. The 2025 version strengthens the UDI application throughout the product lifecycle:
- Each medical device must be assigned a unique UDI code, including product and production identifiers, compliant with global UDI databases (such as the US GUDID and the EU EUDAMED).
- The UDI must be displayed on the product’s smallest saleable packaging and instructions for use, and for implants, the UDI must be embedded in the device itself.
- A traceability system must be established to track the device from raw materials through production, storage, logistics, clinical use, and disposal. Traceability data must be retained for at least 10 years to meet regulatory traceability requirements.
With the acceleration of the digital transformation in the medical device industry, ISO 13485:2025 adds several clauses on electronic records and data security to align with global digital regulatory requirements:
- Electronic records must be stored in tamper-proof formats (such as blockchain technology), with access control and operation logs recorded for at least 10 years, in compliance with FDA 21 CFR Part 11 and EU Annex 11.
- For AI medical devices and medical software, a data security management system must be in place to prevent data breaches, protect patient privacy, and ensure compliance with GDPR and other data protection laws.
- A data backup and emergency recovery mechanism must be implemented to ensure that data is not lost and can be restored in the event of an emergency.
ISO 13485 is not only applicable to medical device manufacturers but covers the entire medical device industry chain. Any organization involved in the design, development, production, procurement, sales, services, sterilization, warehousing, or disposal of medical devices must comply with the requirements of this standard. The specific applicable entities include:
These include manufacturers of active medical devices (such as ventilators, monitors), passive medical devices (such as syringes, joint implants), in vitro diagnostic reagents (such as nucleic acid test kits, glucose test strips), medical software and AI-based medical devices (such as telemedicine software, AI imaging diagnostic equipment), and 3D printed medical devices. These entities are the core group to which the standard applies and must fully meet all the requirements of the standard.
This includes suppliers of medical device raw materials (such as medical plastics, metal materials), component suppliers (such as sensors, chips), packaging material suppliers, sterilization service providers, and logistics companies. These entities must meet the requirements in the standard related to supply chain management, traceability, and sterile control, and cooperate with core manufacturers to complete compliance audits.
This includes medical device distributors, agents, after-sales service organizations, clinical institutions (such as hospitals and clinics), and medical device disposal and recycling organizations. These entities must meet the traceability, after-sales service, and adverse event reporting requirements in the standard to ensure compliance and safety in the distribution and use of products.
ISO 13485 also applies to contract manufacturing and contract research scenarios, where the contracting party and the contractor must clearly define their respective quality responsibilities. The contractor must have a quality management system that meets the standard, and the contracting party must audit and manage the contractor’s quality system. Additionally, the standard applies to medical device export companies, which must establish a quality system based on ISO 13485 requirements to meet the regulatory entry requirements of the target market.
ISO 13485 certification is voluntary, but as it is considered a prerequisite for market access in most countries, many companies proactively apply for certification. The certification process is divided into six steps and must align with the standard’s requirements to ensure the effective implementation of the system:
The company must first define the scope of certification (such as product categories and involved processes), review the regulatory requirements of the target market (such as NMPA in China, MDR in the EU, QSR 820 in the US), and establish a quality system building plan based on the latest version of ISO 13485 (2025). The company should also form a specialized team, define roles and responsibilities, and conduct training on the standard and regulations to ensure all relevant personnel understand the core requirements of the standard.
The company must compile quality system documents based on the standard and the company’s actual situation. These documents typically include the quality manual, procedures, work instructions, and forms. The documents must cover all clauses of the standard and be tailored to the company’s product characteristics and production processes. Once the documentation is completed, the quality system must be implemented, with a minimum operational cycle of 3 months. During this time, complete operation records (such as production records, inspection records, risk assessment records, and management review records) must be retained to verify the feasibility and effectiveness of the system.
The company must form an internal audit team and conduct internal audits based on ISO 13485 requirements. The focus of the audit is to assess the compliance of the system documents, the effectiveness of system implementation, and identify any compliance gaps or quality issues. After identifying issues, corrective and preventive actions must be developed. Once corrective actions are implemented, audit reports and corrective records should be retained to ensure the system meets the certification audit requirements.
The highest management team of the company must conduct a management review, assessing the effectiveness, suitability, and adequacy of the quality system based on internal audit results, system operation data, customer feedback, regulatory updates, and risk assessment results. The company should develop a system improvement plan to address identified deficiencies, and management review reports should be retained to ensure the system always aligns with the company’s development and regulatory requirements.
The company selects a qualified certification body (such as TÜV, SGS, or CQC in China) and submits a certification application. The application must include quality system documents, system operation records, internal audit reports, management review reports, and product-related materials (such as product registration certificates, inspection reports). The certification body will review the application materials and, if approved, arrange for an on-site audit.
Certification body auditors will visit the company’s site to check the actual implementation of the quality system, focusing on areas such as the production workshop, inspection laboratory, and warehouse. Auditors will review operational records and interview relevant personnel to determine if the system complies with ISO 13485. If non-conformities are found during the on-site audit, the company must correct them within a specified time and submit a correction report with verification materials. Once the corrective actions are verified as effective, the certification body will issue the ISO 13485 certification. The certificate is valid for 3 years, during which annual surveillance audits are required. After 3 years, a recertification process must be conducted.
Practitioners often confuse ISO 13485, ISO 9001, and GMP, as all three are related to quality control. However, there are significant differences in their positioning, scope, and core requirements, as well as some connections between them. It is essential to clearly distinguish between them:
Connection:
Both ISO 13485 and ISO 9001 are based on the PDCA cycle (Plan-Do-Check-Act), aiming to improve quality management through standardized processes. ISO 13485 borrows its foundational framework from ISO 9001, and companies that have obtained ISO 13485 certification can reuse some of their system documents to reduce the workload of ISO 9001 certification.
Differences:
- Positioning: ISO 9001 is a general quality management standard applicable to all industries, with a core focus on “customer satisfaction and continuous improvement.” ISO 13485 is a specialized standard for medical devices, with its focus on “regulatory compliance and risk control.”
- Clause Differences: ISO 13485 eliminates clauses unrelated to the medical device industry that are present in ISO 9001 and adds specialized clauses for risk management, UDI traceability, sterile control, etc., with stricter requirements for regulatory compliance.
- Applicable Scenarios: ISO 9001 can be used as a general tool for improving quality management, while ISO 13485 is a critical prerequisite for market access for medical device companies.
Connection:
GMP (Good Manufacturing Practice) in the pharmaceutical industry has a counterpart in the medical device industry—medical device GMP. Both standards aim to ensure “product quality and safety,” emphasizing process control, sterile control, and traceability in production. ISO 13485 certification can serve as an important reference for GMP inspections, and many companies will establish a quality system that meets the requirements of both standards simultaneously.
Differences:
- Nature: GMP is a mandatory requirement, and medical device manufacturers must comply with GMP standards to obtain a production license. ISO 13485 is a voluntary certification, though it is a prerequisite for market access in many countries.
- Scope: GMP focuses on quality control during production, while ISO 13485 covers the entire lifecycle, including design, development, procurement, sales, and services.
- Focus of Control: GMP focuses more on compliance in the production process, with detailed controls over production environment, equipment, and personnel. ISO 13485, on the other hand, emphasizes the overall integrity of the system, with a focus on risk control and regulatory collaboration throughout the entire chain.
In many countries and regions, ISO 13485 certification is a prerequisite for market entry for medical devices. For example, the EU MDR requires that medical device manufacturers implement a quality system compliant with ISO 13485 to obtain CE certification. The FDA recognizes ISO 13485 as an equivalent standard to QSR 820, simplifying the FDA registration process. In China, the NMPA does not mandate ISO 13485 certification, but it is a key factor in medical device registration and production license checks. Obtaining certification increases the likelihood of passing these checks. Additionally, ISO 13485 is a globally recognized standard, and certification reduces compliance costs for companies entering international markets.
ISO 13485 centers on lifecycle risk management, helping companies identify, assess, and control quality and compliance risks across the entire product lifecycle. It reduces deviations and non-conforming products during production, minimizes rework and scrap costs, and mitigates the risk of penalties, product recalls, and legal disputes arising from regulatory non-compliance, especially for high-risk products like AI medical devices and implants.
ISO 13485 certification proves the company’s quality commitment and regulatory compliance, enhancing its credibility in the medical device market. It improves the company’s brand recognition, attracts high-quality customers, and increases confidence in the company’s products. Particularly for global enterprises, ISO 13485 is often regarded as a “global business passport,” increasing business opportunities.
Through the implementation of ISO 13485, companies optimize their internal processes, improve efficiency, reduce waste, and enhance product quality. The establishment of a compliance-driven quality management system enables continuous improvement and problem-solving mechanisms to help companies optimize design, development, production, and service processes.
Many companies face numerous challenges during the establishment and certification process of ISO 13485, especially with the new requirements in the 2025 version. It is essential to develop targeted strategies to ensure effective implementation of the system:
The regulatory requirements for medical devices differ significantly across regions, including the EU MDR, US FDA QSR 820, and China’s NMPA regulations. Companies find it challenging to build a quality system that meets the global regulatory requirements, especially with the new digitalization and AI-related clauses in the 2025 version, which require alignment with multiple national regulatory frameworks.
Strategy:
Form a professional regulatory team to identify the differences between regulations in target markets. Develop differentiated compliance solutions that integrate the requirements of the ISO 13485:2025 standard. Collaborate with third-party consulting firms for regulatory adaptation assessments and timely updates of quality system documents. Establish a regulatory update alert mechanism to track global regulatory changes and optimize the quality system accordingly.
With the rapid development of AI and 3D printed medical devices, companies struggle to maintain adequate quality control over these new technologies, failing to meet the additional requirements in the 2025 version, such as algorithm validation, parameter recording, and data security.
Strategy:
Introduce specialized technical teams to establish control processes for new technologies, such as AI algorithm validation procedures and 3D printing parameter control processes. Work with third-party organizations to carry out algorithm validation and biocompatibility testing to ensure compliance. Strengthen technical personnel training to improve their ability to control the quality of new technologies and adapt to the new requirements in the standard.
Some companies focus only on their own compliance, neglecting control over the upstream and downstream parts of the supply chain. Insufficient supplier qualification audits and incomplete traceability data make full-chain traceability impossible, especially with the stricter supply chain audit requirements introduced in the 2025 version.
Strategy:
Establish a tiered supplier management system to conduct differentiated audits based on the risk level of suppliers, with high-risk suppliers requiring on-site audits. Sign quality agreements with suppliers to clarify traceability and compliance requirements, and require suppliers to provide ISO 13485 certification or compliance proofs. Build a supply chain traceability platform to enable data communication with suppliers, ensuring full-chain traceability of raw materials and components.
Some companies only create superficially compliant system documents to obtain certification, without integrating the system’s requirements into actual production operations. As a result, the system becomes a formality and fails to effectively manage risks, often leading to non-conformities during audits.
Strategy:
Build the system to align with actual production processes to avoid “template-based” implementation. Provide comprehensive training for all staff, ensuring that each position understands its specific system requirements. Establish a regular internal system audit mechanism to periodically review the system’s operation, identify any discrepancies with actual practices, and address them in a timely manner to improve the system’s effectiveness.
ISO 13485, as the quality management system standard for the medical device industry, its core value lies not in simply “obtaining a certificate,” but in achieving regulatory compliance, risk control, and quality stability through a standardized full-chain management system, ensuring patient health and safety. From the 1996 version’s basic framework to the 2025 version’s digital and intelligent upgrades, ISO 13485 has always kept pace with industry developments, becoming a core support for global medical device companies’ compliant operations and market competition.
At PCBCool, we understand the critical importance of complying with ISO 13485 for medical device manufacturers. Partnering with a trusted ISO 13485-certified manufacturer like us for your medical electronics solutions ensures not only regulatory compliance but also enhances the quality and reliability of your products. From the design phase to manufacturing, assembly, and final product integration, PCBCool offers comprehensive end-to-end solutions. Our extensive experience and commitment to precision allow us to deliver high-quality, compliant medical devices that meet the evolving standards of the industry. Whether you’re developing new devices or scaling production, our expertise will support you every step of the way.