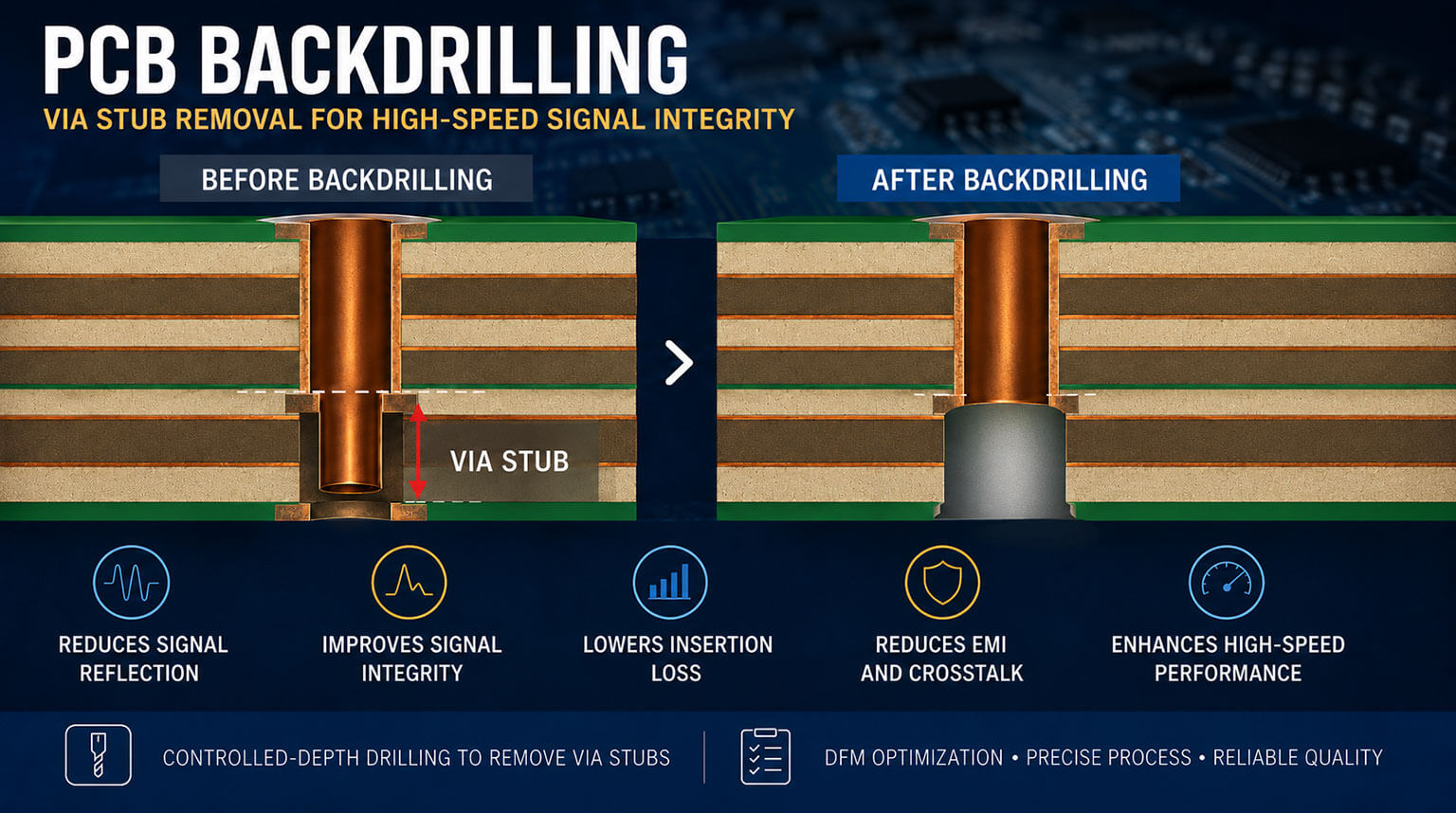
En la industria de dispositivos médicos, la calidad y la seguridad son prioridades innegociables. La norma ISO 13485, un sistema de gestión de calidad (SGC) dedicado para este sector, sirve como la guía principal para el cumplimiento, el acceso al mercado y la gestión de riesgos a nivel mundial. Ya sean dispositivos activos, implantes pasivos, reactivos de diagnóstico in vitro o software médico, la ISO 13485 abarca todo el ciclo de vida del producto, desde el diseño y desarrollo, pasando por la fabricación, hasta la trazabilidad posterior a la comercialización. Define el “lenguaje universal” del control de calidad dentro de la industria de dispositivos médicos.
este artículo de PCBCool desglosará a fondo la norma ISO 13485 desde varias dimensiones: el origen de la norma, su evolución, los requisitos fundamentales, el alcance de aplicación y el valor de la certificación. Esto ayudará a los profesionales a obtener una comprensión más profunda de su lógica central y los puntos clave de implementación.
La norma ISO 13485, titulada oficialmente Dispositivos médicos — Sistemas de gestión de la calidad — Requisitos para fines reglamentarios, es una norma especializada desarrollada por la Organización Internacional de Normalización (ISO) bajo su comité técnico de dispositivos médicos (ISO/TC 210). El enfoque principal de la ISO 13485 es “el cumplimiento normativo en el núcleo y la gestión de riesgos como herramienta”, proporcionando un marco práctico para la gestión de la calidad en toda la cadena de la industria de dispositivos médicos para garantizar que los productos cumplan con los requisitos reglamentarios mundiales y las demandas de seguridad y eficacia clínica.
A diferencia de las normas de gestión de calidad generales como la ISO 9001, la ISO 13485 resta importancia al requisito universal de “mejora continua” y pone un mayor énfasis en el “mantenimiento de la efectividad del sistema” y la “conformidad regulatoria” obligatorios. No es una norma de calidad independiente, sino que necesita integrar profundamente los diversos marcos regulatorios nacionales para dispositivos médicos (como las regulaciones de la NMPA de China, el MDR de la UE y el QSR 820 de la FDA de EE. UU.). El objetivo principal es estandarizar los procesos de gestión para reducir los riesgos de seguridad a lo largo del ciclo de vida del producto, garantizando la salud y seguridad de los pacientes y usuarios.
Es importante señalar que la norma ISO 13485 es un sistema de normas independiente. Aunque se basa en el concepto del ciclo PDCA (Planificar-Hacer-Verificar-Actuar) de la ISO 9001, introduce numerosos requisitos específicos adaptados a las necesidades únicas de la industria de dispositivos médicos, cubriendo áreas críticas como la gestión de riesgos, el control de la esterilización, la trazabilidad UDI y la gestión de eventos adversos. Esto la convierte en un “pasaporte” esencial para las empresas de dispositivos médicos que buscan ingresar a los mercados globales.
El desarrollo de la norma ISO 13485 siempre ha estado sincronizado con las actualizaciones regulatorias globales en el sector de los dispositivos médicos y las innovaciones tecnológicas. La norma ha pasado por tres revisiones importantes, ampliando progresivamente su cobertura de toda la cadena industrial y las nuevas tecnologías, lo que ha dado lugar a la versión actual de la norma:
En 1996, la ISO publicó la primera edición de la norma ISO 13485, que reemplazó el contenido relevante de la norma ISO 8402 relativo a los dispositivos médicos. El valor central de esta edición fue “establecer un marco especializado para la industria”. Introdujo terminología específica y requisitos básicos para los sistemas de gestión de calidad de dispositivos médicos, alineándose con los marcos regulatorios vigentes en ese momento en Europa y América del Norte. Esta edición proporcionó una base para que la industria superara las limitaciones de las normas generales de calidad. Sin embargo, los términos eran relativamente básicos y el enfoque se centraba principalmente en el control de calidad durante la etapa de fabricación, sin cubrir el ciclo de vida completo ni la gestión de riesgos.
En 2003, la ISO llevó a cabo la primera revisión importante, publicando la ISO 13485:2003. Los cambios clave en esta versión fueron “el fortalecimiento de la compatibilidad reglamentaria y los fundamentos de la gestión de riesgos”. Esta edición definió explícitamente el propósito central de los “requisitos reglamentarios” e integró los fundamentos de la gestión de riesgos. También detalló requisitos específicos para el diseño y desarrollo, la fabricación y las pruebas, alineándose más estrechamente con la EU MDD (Directiva sobre Productos Sanitarios) y la US FDA QSR 820 en ese momento.
En marzo de 2016, la ISO publicó la ISO 13485:2016, que entró en vigor en 2017. Las principales actualizaciones de esta edición se centraron en la “gestión del ciclo de vida y la armonización regulatoria global”. Esta versión amplió la penetración de la gestión de riesgos en todo el proceso, exigiendo la integración de la ISO 14971 (la norma de gestión de riesgos para dispositivos médicos). También añadió requisitos específicos para la gestión de la cadena de suministro, la trazabilidad del producto y el control del software. Esta versión se alineó más estrechamente con las tendencias regulatorias globales y eliminó cláusulas redundantes que se solapaban con la ISO 9001.
El 9 de mayo de 2025, la ISO lanzará oficialmente la ISO 13485:2025, marcando la entrada de la gestión de la calidad de los dispositivos médicos en una nueva era de “digitalización, inteligencia y cumplimiento de cadena completa”. Esta revisión es una actualización integral de la edición de 2016 y se centra en cuatro áreas clave:
- La adición de cláusulas sobre ética y seguridad de datos para dispositivos médicos basados en IA, adaptándose a nuevas tecnologías como el Software como Dispositivo Médico (SaMD) y dispositivos médicos impresos en 3D.
- Fortalecimiento del control de registros electrónicos, incluyendo el uso de blockchain y otras tecnologías para prevenir la manipulación de datos, en cumplimiento con la FDA 21 CFR Parte 11 y el Anexo 11 de la UE.
- Gestión de la cadena de suministro mejorada, implementando auditorías por niveles e inspecciones obligatorias in situ, extendiendo el control a los proveedores de materias primas.
- Ampliando la trazabilidad a 10 años y mejorando los mecanismos de vigilancia posterior a la comercialización (PMS).
La estructura de cláusulas de la ISO 13485 se centra en el ’cumplimiento normativo, control de riesgos y trazabilidad“, abarcando 8 capítulos principales. Se puede dividir en cinco módulos de gestión centrales y cuatro requisitos de control especiales, equilibrando la aplicabilidad general con las necesidades específicas de la industria. La versión de 2025 fortalece aún más la digitalización y la adaptabilidad a las nuevas tecnologías, convirtiéndose en un foco clave para las auditorías.
El requisito fundamental es que las organizaciones establezcan un marco claro de gestión de la calidad, definan funciones y responsabilidades, y garanticen el compromiso de la alta dirección con el cumplimiento normativo y la asignación de recursos.
La versión de 2025 agrega un nuevo requisito: el CEO debe presidir las revisiones trimestrales del cumplimiento normativo, incluyendo evaluaciones del cumplimiento de la ética de la IA. También exige que las empresas establezcan políticas y objetivos de calidad que sean medibles, procesables y directamente vinculados a los riesgos de seguridad del producto y a los requisitos normativos. Además, se debe establecer un mecanismo de revisión por la dirección para evaluar regularmente la eficacia del sistema, abordando rápidamente los riesgos de cumplimiento y los problemas de calidad.
Este módulo se centra en los cinco recursos principales: personas, equipos, materiales, métodos y entorno.
Para el personal, los puestos clave (como inspección de esterilización, evaluación de riesgos y revisión de productos) deben ser ocupados por personas con las cualificaciones reconocidas por la NMPA (Administración Nacional de Productos Médicos de China), y se debe realizar capacitación periódica sobre regulaciones de dispositivos médicos y habilidades de control de calidad, manteniendo registros completos.
En cuanto a la infraestructura, los equipos de producción e inspección deben calibrarse y mantenerse regularmente para garantizar la precisión. Los equipos especializados (como la impresión 3D y las herramientas de detección de IA) deben contar con un proceso de verificación digital. Para el entorno de trabajo, la fabricación de productos estériles debe realizarse en salas limpias certificadas, con la versión 2025 que exige el monitoreo en tiempo real de la temperatura y la humedad y la carga de datos basada en la nube para la trazabilidad. Además, debe existir un mecanismo de aseguramiento de recursos para garantizar que los fondos, la tecnología y el personal necesarios estén disponibles para el control de calidad, evitando riesgos de calidad debido a recursos insuficientes.
Este es el módulo central de la ISO 13485, que abarca todo el proceso desde el diseño y desarrollo hasta la compra, la fabricación y los servicios posventa, con requisitos de cumplimiento específicos para cada etapa:
- Diseño y Desarrollo Se debe establecer un proceso integral de control de diseño, especificando las entradas de diseño (incluidos los objetivos de seguridad del paciente), las salidas, las revisiones y los requisitos de validación/verificación. La versión 2025 agrega requisitos para la validación de algoritmos de IA y registros de parámetros de impresión 3D. La documentación de control de diseño (DHF) debe incluir datos sobre pruebas de biocompatibilidad y validación de algoritmos para garantizar que los diseños cumplan con las normas reglamentarias y de seguridad.
- Compras Los proveedores deben ser evaluados y controlados mediante evaluaciones por niveles, y los proveedores de alto riesgo deben someterse a auditorías en el sitio. Los proveedores deben demostrar la trazabilidad UDI (Identificación Única de Dispositivo) y firmar acuerdos de calidad que definan los estándares de calidad de los materiales y los requisitos de trazabilidad. Los implantes deben rastrear los materiales hasta el nivel de materia prima.
- Producción Se deben desarrollar procedimientos operativos estándar (POE) para controlar los parámetros del proceso. Se debe implementar el “seguimiento de unidades individuales” (cobertura completa de UDI) para eliminar desviaciones durante la producción. Para productos estériles, los procesos de esterilización deben controlarse estrictamente, conservando los registros completos de esterilización.
- Servicios: Se debe establecer un proceso de servicio post-mercado para manejar las quejas de los clientes y los problemas de calidad de manera oportuna. Se deben recopilar comentarios para la vigilancia post-mercado, y la versión de 2025 requerirá que los registros de servicio se vinculen al sistema de trazabilidad del producto para una trazabilidad completa del ciclo de vida.
Las empresas deben establecer mecanismos de seguimiento y análisis para rastrear continuamente la eficacia del sistema de gestión de la calidad, la calidad del producto y el cumplimiento normativo:
- Las inspecciones deben realizarse en cada lote de productos, y los registros de inspección deben conservarse durante al menos cinco años.
- Debe existir un sistema para la recopilación y notificación de eventos adversos, debiendo estos eventos adversos ser notificados en un plazo de 24 horas a las plataformas regulatorias.
- El análisis de datos debe usarse para identificar riesgos de calidad y cumplimiento, y los informes analíticos deben servir de base para las acciones correctivas.
- Se debe implementar un sistema de acciones correctivas y preventivas (CAPA) para abordar los problemas identificados, con responsabilidades, plazos y requisitos de verificación claros. La versión 2025 requiere que el análisis de las tendencias de eventos adversos se incluya en las revisiones de gestión para mejorar las capacidades de advertencia de riesgos.
Las empresas deben establecer un sistema de gestión de retiradas de productos, definiendo sus procesos, responsabilidades y planes de emergencia. Se debe realizar un ejercicio de retirada simulado al menos una vez al año, e incluir las tasas de retirada en las revisiones anuales de la dirección. Además, las mejoras continuas del sistema de calidad deben basarse en las revisiones de la dirección, el análisis de datos y la retroalimentación del cliente, asegurando un ciclo de “cumplimiento-optimización-recumplimiento”. A diferencia de la ISO 9001, el enfoque aquí está más en mantener el cumplimiento normativo que en la mejora del rendimiento. El objetivo principal es garantizar que el sistema de gestión de calidad se mantenga actualizado con los últimos requisitos normativos, minimizando los riesgos de incumplimiento.
Desde la versión de 2016, la norma ISO 13485 ha exigido que las empresas integren la norma ISO 14971 (la norma de gestión de riesgos de productos sanitarios) en sus sistemas de gestión de calidad. La versión de 2025 refina aún más los requisitos para el control de riesgos. La lógica fundamental es la “gestión de riesgos del ciclo de vida”, que abarca la identificación, evaluación y control de riesgos desde el diseño hasta la producción y la vigilancia posterior a la comercialización. Las medidas de control de riesgos deben ser documentadas, verificables y trazables.
Para los dispositivos basados en IA, la versión de 2025 introduce el requisito de “clasificación de riesgo de algoritmos”, donde los algoritmos de alto riesgo deben someterse a una verificación por parte de terceros. Para los dispositivos implantables, se requiere un seguimiento de riesgos a largo plazo de hasta 10 años, manteniendo los datos de riesgo.
Para los dispositivos médicos estériles (como jeringuillas, implantes articulares, apósitos estériles), la norma ISO 13485 establece requisitos de control específicos. La versión de 2025 mejora el control sobre los entornos limpios y los procesos de esterilización:
- Las salas limpias deben cumplir con las normas ISO 14644, y los niveles de limpieza deben definirse según las clasificaciones de riesgo del producto. Se requiere el monitoreo en tiempo real de partículas suspendidas y microorganismos, con los datos cargados en plataformas regulatorias.
- Los procesos de esterilización deben utilizar métodos validados (como vapor o irradiación), conservando los parámetros de esterilización y los informes de validación para garantizar la repetibilidad y trazabilidad del proceso de esterilización.
- Debe existir un sistema de barrera estéril para prevenir la contaminación durante el almacenamiento y el transporte, y los materiales de envasado para productos estériles deben someterse a pruebas de biocompatibilidad.
La Identificación Única de Dispositivos (UDI) es fundamental para el sistema de trazabilidad de la norma ISO 13485. La versión de 2025 refuerza la aplicación de la UDI a lo largo de todo el ciclo de vida del producto:
- Cada dispositivo médico debe tener asignado un código UDI único, que incluya identificadores de producto y producción, cumpliendo con las bases de datos globales de UDI (como la GUDID de EE. UU. y la EUDAMED de la UE).
- La DUC debe mostrarse en el embalaje vendible más pequeño del producto e instrucciones de uso, y para los implantes, la DUC debe estar incrustada en el propio dispositivo.
- Se debe establecer un sistema de trazabilidad para rastrear el dispositivo desde las materias primas hasta la producción, almacenamiento, logística, uso clínico y eliminación. Los datos de trazabilidad deben conservarse durante al menos 10 años para cumplir con los requisitos regulatorios de trazabilidad.
Con la aceleración de la transformación digital en la industria de dispositivos médicos, la norma ISO 13485:2025 añade varias cláusulas sobre registros electrónicos y seguridad de datos para alinearse con los requisitos regulatorios digitales globales:
- Los registros electrónicos deben almacenarse en formatos a prueba de manipulaciones (como la tecnología blockchain), con control de acceso y registros de operaciones para al menos 10 años, cumpliendo con la FDA 21 CFR Parte 11 y el Anexo 11 de la UE.
- Para dispositivos médicos e software médico basados en IA, debe existir un sistema de gestión de seguridad de datos para prevenir violaciones de datos, proteger la privacidad del paciente y garantizar el cumplimiento del RGPD y otras leyes de protección de datos.
- Se debe implementar un mecanismo de respaldo de datos y recuperación de emergencias para garantizar que los datos no se pierdan y puedan ser restaurados en caso de emergencia.
La norma ISO 13485 no solo se aplica a los fabricantes de dispositivos médicos, sino que abarca toda la cadena de la industria de dispositivos médicos. Cualquier organización involucrada en el diseño, desarrollo, producción, adquisición, venta, servicio, esterilización, almacenamiento o eliminación de dispositivos médicos debe cumplir con los requisitos de esta norma. Las entidades específicamente aplicables incluyen:
Estos incluyen fabricantes de dispositivos médicos activos (como ventiladores, monitores), dispositivos médicos pasivos (como jeringas, implantes articulares), reactivos de diagnóstico in vitro (como kits de pruebas de ácidos nucleicos, tiras reactivas para glucosa), software médico y dispositivos médicos basados en IA (como software de telemedicina, equipos de diagnóstico por imagen con IA), y dispositivos médicos impresos en 3D. Estas entidades constituyen el grupo central al que se aplica la norma y deben cumplir íntegramente todos los requisitos de la misma.
Esto incluye proveedores de materias primas para dispositivos médicos (como plásticos médicos, materiales metálicos), proveedores de componentes (como sensores, chips), proveedores de materiales de embalaje, proveedores de servicios de esterilización y empresas de logística. Estas entidades deben cumplir los requisitos de la norma relacionados con la gestión de la cadena de suministro, la trazabilidad y el control estéril, y cooperar con los fabricantes principales para completar las auditorías de cumplimiento.
Esto incluye distribuidores de dispositivos médicos, agentes, organizaciones de servicio posventa, instituciones clínicas (como hospitales y clínicas) y organizaciones de eliminación y reciclaje de dispositivos médicos. Estas entidades deben cumplir con los requisitos de trazabilidad, servicio posventa y notificación de eventos adversos en la norma para garantizar el cumplimiento y la seguridad en la distribución y el uso de los productos.
La norma ISO 13485 también se aplica a escenarios de fabricación y investigación por contrato, donde la parte contratante y el contratista deben definir claramente sus respectivas responsabilidades de calidad. El contratista debe contar con un sistema de gestión de la calidad que cumpla con la norma, y la parte contratante debe auditar y gestionar el sistema de calidad del contratista. Además, la norma se aplica a las empresas exportadoras de dispositivos médicos, que deben establecer un sistema de calidad basado en los requisitos de la ISO 13485 para cumplir con los requisitos reglamentarios de entrada del mercado de destino.
La certificación ISO 13485 es voluntaria, pero como se considera un requisito previo para el acceso al mercado en la mayoría de los países, muchas empresas solicitan la certificación de forma proactiva. El proceso de certificación se divide en seis pasos y debe alinearse con los requisitos de la norma para garantizar la implementación efectiva del sistema:
La empresa debe primero definir el alcance de la certificación (como categorías de productos y procesos involucrados), revisar los requisitos regulatorios del mercado objetivo (como NMPA en China, MDR en la UE, QSR 820 en EE. UU.) y establecer un plan de construcción del sistema de calidad basado en la última versión de ISO 13485 (2025). La empresa también debe formar un equipo especializado, definir roles y responsabilidades, y realizar capacitación sobre la norma y las regulaciones para asegurar que todo el personal relevante comprenda los requisitos centrales de la norma.
La empresa debe compilar los documentos del sistema de calidad basándose en la norma y la situación real de la empresa. Estos documentos suelen incluir el manual de calidad, los procedimientos, las instrucciones de trabajo y los formularios. Los documentos deben abarcar todas las cláusulas de la norma y adaptarse a las características del producto y a los procesos de producción de la empresa. Una vez completada la documentación, se debe implementar el sistema de calidad, con un ciclo operativo mínimo de 3 meses. Durante este tiempo, se deben conservar los registros completos de operación (como registros de producción, registros de inspección, registros de evaluación de riesgos y registros de revisión de la dirección) para verificar la viabilidad y eficacia del sistema.
La empresa debe formar un equipo de auditoría interna y realizar auditorías internas basadas en los requisitos de la norma ISO 13485. El enfoque de la auditoría es evaluar el cumplimiento de los documentos del sistema, la efectividad de la implementación del sistema e identificar cualquier incumplimiento o problema de calidad. Después de identificar los problemas, se deben desarrollar acciones correctivas y preventivas. Una vez implementadas las acciones correctivas, se deben conservar los informes de auditoría y los registros correctivos para garantizar que el sistema cumpla con los requisitos de la auditoría de certificación.
El equipo directivo de mayor nivel de la empresa debe realizar una revisión por la dirección, evaluando la eficacia, idoneidad y adecuación del sistema de gestión de calidad basándose en los resultados de las auditorías internas, datos de operación del sistema, comentarios de los clientes, actualizaciones regulatorias y resultados de la evaluación de riesgos. La empresa debe desarrollar un plan de mejora del sistema para abordar las deficiencias identificadas, y se deben conservar los informes de revisión por la dirección para garantizar que el sistema se alinee siempre con el desarrollo de la empresa y los requisitos reglamentarios.
La empresa selecciona un organismo de certificación cualificado (como TÜV, SGS o CQC en China) y presenta una solicitud de certificación. La solicitud debe incluir documentos del sistema de calidad, registros de operación del sistema, informes de auditoría interna, informes de revisión de la dirección y materiales relacionados con el producto (como certificados de registro de producto, informes de inspección). El organismo de certificación revisará los materiales de la solicitud y, si se aprueban, organizará una auditoría in situ.
Los auditores del organismo de certificación visitarán las instalaciones de la empresa para verificar la implementación real del sistema de calidad, centrándose en áreas como el taller de producción, el laboratorio de inspección y el almacén. Los auditores revisarán los registros operativos y entrevistarán al personal relevante para determinar si el sistema cumple con la norma ISO 13485. Si se encuentran no conformidades durante la auditoría in situ, la empresa deberá corregirlas dentro de un plazo especificado y presentar un informe de corrección con materiales de verificación. Una vez que se verifique la eficacia de las acciones correctivas, el organismo de certificación emitirá la certificación ISO 13485. El certificado tiene una validez de 3 años, durante los cuales se requieren auditorías de seguimiento anuales. Después de 3 años, se debe realizar un proceso de recertificación.
Los profesionales a menudo confunden la ISO 13485, la ISO 9001 y las GMP, ya que las tres están relacionadas con el control de calidad. Sin embargo, existen diferencias significativas en su posicionamiento, alcance y requisitos centrales, así como algunas conexiones entre ellas. Es esencial distinguirlas claramente:
Conexión:
Tanto la ISO 13485 como la ISO 9001 se basan en el ciclo PDCA (Planificar-Hacer-Verificar-Actuar), con el objetivo de mejorar la gestión de la calidad a través de procesos estandarizados. La ISO 13485 toma su marco fundamental de la ISO 9001, y las empresas que han obtenido la certificación ISO 13485 pueden reutilizar algunos de los documentos de su sistema para reducir la carga de trabajo de la certificación ISO 9001.
Diferencias:
- Posicionamiento: ISO 9001 es un estándar general de gestión de calidad aplicable a todas las industrias, con un enfoque central en la “satisfacción del cliente y la mejora continua”. ISO 13485 es un estándar especializado para dispositivos médicos, con su enfoque en el “cumplimiento normativo y el control de riesgos”.”
- Diferencias de cláusulas: La norma ISO 13485 elimina las cláusulas no relacionadas con la industria de dispositivos médicos que están presentes en la ISO 9001 y agrega cláusulas especializadas para gestión de riesgos, trazabilidad UDI, control estéril, etc., con requisitos más estrictos para el cumplimiento normativo.
- Escenarios aplicables: La ISO 9001 se puede utilizar como una herramienta general para mejorar la gestión de la calidad, mientras que la ISO 13485 es un requisito previo crítico para el acceso al mercado de las empresas de dispositivos médicos.
Conexión:
Las GMP (Buenas Prácticas de Fabricación) en la industria farmacéutica tienen un homólogo en la industria de dispositivos médicos: las GMP de dispositivos médicos. Ambas normas tienen como objetivo garantizar la “calidad y seguridad del producto”, enfatizando el control de procesos, el control estéril y la trazabilidad en la producción. La certificación ISO 13485 puede servir como una referencia importante para las inspecciones GMP, y muchas empresas establecerán un sistema de calidad que cumpla con los requisitos de ambas normas simultáneamente.
Diferencias:
- Naturaleza: Las BPF son un requisito obligatorio y los fabricantes de dispositivos médicos deben cumplir con las normas de BPF para obtener una licencia de producción. La ISO 13485 es una certificación voluntaria, aunque es un requisito previo para el acceso al mercado en muchos países.
- Alcance GMP se enfoca en el control de calidad durante la producción, mientras que ISO 13485 cubre todo el ciclo de vida, incluyendo diseño, desarrollo, adquisición, ventas y servicios.
- Locus de Control Las GMP se centran más en el cumplimiento en el proceso de producción, con controles detallados sobre el entorno de producción, el equipo y el personal. La ISO 13485, por otro lado, enfatiza la integridad general del sistema, con un enfoque en el control de riesgos y la colaboración regulatoria a lo largo de toda la cadena.
En muchos países y regiones, la certificación ISO 13485 es un requisito previo para la comercialización de productos sanitarios. Por ejemplo, el Reglamento sobre productos sanitarios (MDR) de la UE exige que los fabricantes de productos sanitarios implementen un sistema de calidad conforme a la norma ISO 13485 para obtener la marca CE. La FDA reconoce la norma ISO 13485 como equivalente a la norma QSR 820, lo que simplifica el proceso de registro ante la FDA. En China, la NMPA no exige la certificación ISO 13485, pero es un factor clave en el registro de productos sanitarios y en las comprobaciones de las licencias de producción. Obtener la certificación aumenta las probabilidades de superar estas comprobaciones. Además, la ISO 13485 es una norma reconocida a nivel mundial, y la certificación reduce los costes de cumplimiento normativo para las empresas que se introducen en los mercados internacionales.
La norma ISO 13485 se centra en la gestión de riesgos a lo largo del ciclo de vida, ayudando a las empresas a identificar, evaluar y controlar los riesgos relacionados con la calidad y el cumplimiento normativo a lo largo de todo el ciclo de vida del producto. Reduce las desviaciones y los productos no conformes durante la producción, minimiza los costes de reelaboración y de desechos, y mitiga el riesgo de sanciones, retiradas de productos y disputas legales derivadas del incumplimiento normativo, especialmente en el caso de productos de alto riesgo, como los dispositivos médicos basados en inteligencia artificial y los implantes.
La certificación ISO 13485 demuestra el compromiso de calidad y el cumplimiento normativo de la empresa, mejorando su credibilidad en el mercado de dispositivos médicos. Mejora el reconocimiento de la marca de la empresa, atrae clientes de alta calidad y aumenta la confianza en los productos de la empresa. Especialmente para las empresas globales, la ISO 13485 a menudo se considera un “pasaporte de negocios global”, que aumenta las oportunidades de negocio.
Mediante la implantación de la norma ISO 13485, las empresas optimizan sus procesos internos, mejoran la eficiencia, reducen el desperdicio y mejoran la calidad de los productos. El establecimiento de un sistema de gestión de la calidad basado en el cumplimiento normativo permite la mejora continua y la creación de mecanismos de resolución de problemas que ayudan a las empresas a optimizar los procesos de diseño, desarrollo, producción y servicio.
Muchas empresas se enfrentan a numerosos retos durante el proceso de implantación y certificación de la norma ISO 13485, especialmente con los nuevos requisitos de la versión de 2025. Es fundamental desarrollar estrategias específicas para garantizar una implementación eficaz del sistema:
Los requisitos normativos para los dispositivos médicos difieren significativamente entre regiones, incluyendo el MDR de la UE, las QSR 820 de la FDA de EE. UU. y las regulaciones NMPA de China. A las empresas les resulta difícil construir un sistema de calidad que cumpla con los requisitos normativos globales, especialmente con las nuevas cláusulas relacionadas con la digitalización y la IA en la versión de 2025, que requieren la alineación con múltiples marcos regulatorios nacionales.
Estrategia
Formar un equipo regulatorio profesional para identificar las diferencias entre las regulaciones en los mercados objetivo. Desarrollar soluciones de cumplimiento diferenciadas que integren los requisitos de la norma ISO 13485:2025. Colaborar con empresas consultoras externas para evaluaciones de adaptación regulatoria y actualizaciones oportunas de los documentos del sistema de calidad. Establecer un mecanismo de alerta de actualizaciones regulatorias para rastrear los cambios regulatorios globales y optimizar el sistema de calidad en consecuencia.
Con el rápido desarrollo de la IA y los dispositivos médicos impresos en 3D, las empresas luchan por mantener un control de calidad adecuado sobre estas nuevas tecnologías, al no poder cumplir los requisitos adicionales de la versión 2025, como la validación de algoritmos, el registro de parámetros y la seguridad de los datos.
Estrategia
Introduce equipos técnicos especializados para establecer procesos de control para nuevas tecnologías, como procedimientos de validación de algoritmos de IA y procesos de control de parámetros de impresión 3D. Trabajar con organizaciones externas para llevar a cabo la validación de algoritmos y pruebas de biocompatibilidad para garantizar el cumplimiento. Fortalecer la capacitación del personal técnico para mejorar su capacidad de controlar la calidad de las nuevas tecnologías y adaptarse a los nuevos requisitos de la norma.
Algunas empresas se centran únicamente en su propio cumplimiento, descuidando el control de las partes aguas arriba y aguas abajo de la cadena de suministro. Las auditorías insuficientes de cualificación de proveedores y los datos de trazabilidad incompletos hacen imposible la trazabilidad de toda la cadena, especialmente con los requisitos de auditoría de la cadena de suministro más estrictos introducidos en la versión 2025.
Estrategia
Establecer un sistema de gestión de proveedores por niveles para llevar a cabo auditorías diferenciadas en función del nivel de riesgo de los proveedores, exigiendo auditorías in situ a los proveedores de alto riesgo. Firmar acuerdos de calidad con los proveedores para aclarar los requisitos de trazabilidad y cumplimiento, y exigir a los proveedores que presenten la certificación ISO 13485 o pruebas de cumplimiento. Crear una plataforma de trazabilidad de la cadena de suministro que permita la comunicación de datos con los proveedores, garantizando la trazabilidad de las materias primas y los componentes a lo largo de toda la cadena.
Algunas empresas se limitan a elaborar documentación del sistema que cumple con los requisitos de forma meramente superficial para obtener la certificación, sin integrar dichos requisitos en las operaciones de producción reales. Como consecuencia, el sistema se convierte en una mera formalidad y no logra gestionar los riesgos de manera eficaz, lo que a menudo da lugar a incumplimientos durante las auditorías.
Estrategia
Se debe diseñar el sistema de manera que se adapte a los procesos de producción reales, con el fin de evitar una implementación “basada en plantillas”. Se debe impartir una formación exhaustiva a todo el personal, asegurándose de que cada puesto comprenda los requisitos específicos del sistema. Se debe establecer un mecanismo de auditoría interna periódica del sistema para revisar periódicamente su funcionamiento, identificar cualquier discrepancia con las prácticas reales y abordarlas de manera oportuna con el fin de mejorar la eficacia del sistema.
La norma ISO 13485, como estándar de sistemas de gestión de la calidad para el sector de los productos sanitarios, no tiene como objetivo principal la mera “obtención de un certificado”, sino lograr el cumplimiento normativo, el control de riesgos y la estabilidad de la calidad a través de un sistema de gestión estandarizado que abarca toda la cadena de suministro, garantizando así la salud y la seguridad de los pacientes. Desde el marco básico de la versión de 1996 hasta las actualizaciones digitales e inteligentes de la versión de 2025, la norma ISO 13485 siempre ha seguido el ritmo de los avances del sector, convirtiéndose en un pilar fundamental para el cumplimiento normativo y la competitividad en el mercado de las empresas de productos sanitarios a nivel mundial.
En PCBCool, comprendemos la importancia crítica de cumplir con la ISO 13485 para los fabricantes de dispositivos médicos. Asociarse con un fabricante certificado ISO 13485 de confianza como nosotros para su soluciones de electrónica médica no solo garantiza el cumplimiento normativo, sino que también mejora la calidad y la fiabilidad de sus productos. Desde la fase de diseño hasta la fabricación, el montaje y la integración del producto final, PCBCool ofrece soluciones integrales de principio a fin. Nuestra amplia experiencia y nuestro compromiso con la precisión nos permiten suministrar dispositivos médicos de alta calidad que cumplen con la normativa y se ajustan a los estándares en constante evolución del sector. Tanto si está desarrollando nuevos dispositivos como si está ampliando la producción, nuestra experiencia le acompañará en cada paso del camino.