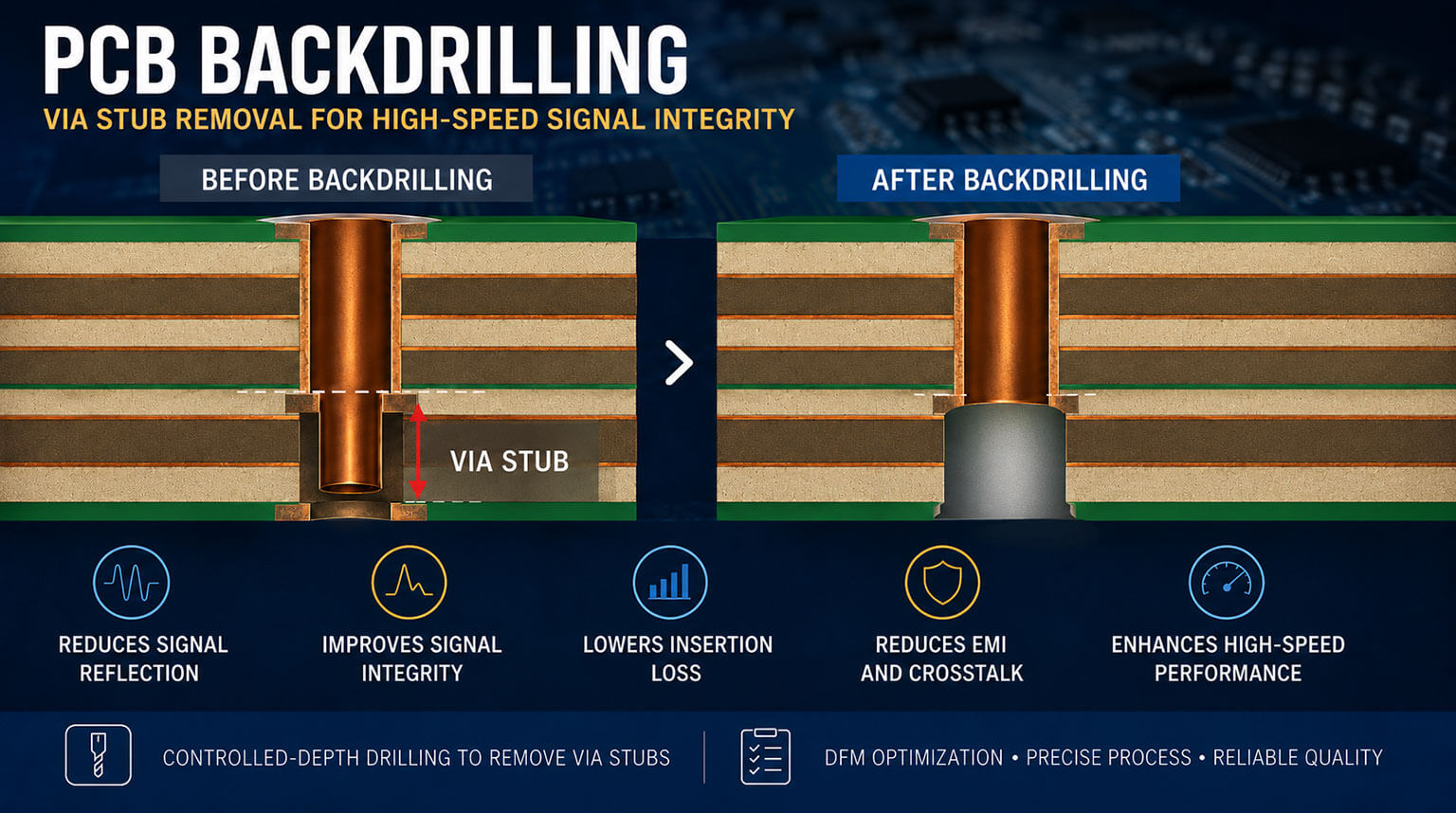
Na indústria de dispositivos médicos, qualidade e segurança são prioridades intransigentes. A ISO 13485, uma norma específica de sistema de gestão da qualidade (SGQ) para este setor, serve como a diretriz central para conformidade, acesso ao mercado e gestão de riscos em todo o mundo. Sejam dispositivos ativos, implantes passivos, reagentes de diagnóstico in vitro ou software médico, a ISO 13485 abrange todo o ciclo de vida do produto – desde o projeto e desenvolvimento, passando pela fabricação, até a rastreabilidade pós-mercado. Ela define a “linguagem universal” do controle de qualidade dentro da indústria de dispositivos médicos.
Este artigo de PCBCool desconstruirá completamente a ISO 13485 em várias dimensões: a origem da norma, sua evolução, os requisitos centrais, o escopo de aplicação e o valor da certificação. Isso ajudará os profissionais a obter uma compreensão mais profunda de sua lógica central e dos principais pontos de implementação.
A ISO 13485, intitulada oficialmente Dispositivos médicos — Sistemas de gestão da qualidade — Requisitos para fins regulamentares, é uma norma especializada desenvolvida pela Organização Internacional de Normalização (ISO) sob seu comitê técnico para dispositivos médicos (ISO/TC 210). O foco principal da ISO 13485 é “conformidade regulatória no núcleo e gestão de riscos como ferramenta”, fornecendo uma estrutura prática para a gestão da qualidade em toda a cadeia da indústria de dispositivos médicos para garantir que os produtos atendam aos requisitos regulatórios globais e às demandas de segurança clínica e eficácia.
Diferentemente de normas gerais de gestão da qualidade, como a ISO 9001, a ISO 13485 minimiza a exigência universal de “melhoria contínua” e enfatiza mais a “manutenção da eficácia do sistema” e a “conformidade regulatória” de forma mandatórias. Ela não é uma norma de qualidade autônoma, mas precisa integrar profundamente os diversos arcabouços regulatórios nacionais para dispositivos médicos (como as regulamentações da NMPA da China, o MDR da UE e o QSR 820 da FDA dos EUA). O objetivo central é padronizar os processos de gestão para reduzir riscos à segurança em todo o ciclo de vida do produto, garantindo a saúde e a segurança de pacientes e usuários.
É importante notar que a ISO 13485 é um sistema de normas independente. Embora se baseie no conceito do ciclo PDCA (Planejar-Fazer-Verificar-Agir) da ISO 9001, introduz numerosos requisitos específicos adaptados às necessidades únicas da indústria de dispositivos médicos, cobrindo áreas críticas como gestão de riscos, controle de esterilização, rastreabilidade UDI e tratamento de eventos adversos. Isso a torna um “passaporte” essencial para empresas de dispositivos médicos que buscam entrar em mercados globais.
O desenvolvimento da ISO 13485 sempre foi sincronizado com as atualizações regulatórias globais no setor de dispositivos médicos e com as inovações tecnológicas. A norma passou por três grandes revisões, expandindo progressivamente sua cobertura de toda a cadeia industrial e de novas tecnologias, o que resultou na versão atual da norma:
Em 1996, a ISO lançou a primeira edição da ISO 13485, substituindo o conteúdo relevante da norma ISO 8402 referente a dispositivos médicos. O valor central desta edição foi “estabelecer um arcabouço especializado para a indústria”. Introduziu terminologia específica e requisitos básicos para sistemas de gestão da qualidade de dispositivos médicos, alinhando-se com os arcabouços regulatórios vigentes na época na Europa e América do Norte. Esta edição forneceu uma base para que a indústria pudesse ir além das limitações das normas gerais de qualidade. No entanto, os termos eram relativamente básicos, e o foco estava predominantemente no controle de qualidade durante a fase de fabricação, sem abranger o ciclo de vida completo ou a gestão de riscos.
Em 2003, a ISO realizou a primeira revisão significativa, lançando a ISO 13485:2003. As principais mudanças nesta versão foram o “fortalecimento da compatibilidade regulatória e dos fundamentos de gerenciamento de riscos”. Esta edição definiu explicitamente o propósito central dos “requisitos regulamentares” e integrou os fundamentos do gerenciamento de riscos. Ela também detalhou requisitos específicos para projeto e desenvolvimento, fabricação e testes, alinhando-se mais estreitamente com a EU MDD (Diretiva de Dispositivos Médicos) e a US FDA QSR 820 na época.
Em março de 2016, a ISO publicou a ISO 13485:2016, que entrou em vigor em 2017. As principais atualizações nesta edição foram focadas no “gerenciamento do ciclo de vida e harmonização regulatória global”. Esta versão expandiu a aplicação do gerenciamento de riscos em todo o processo, exigindo a integração da ISO 14971 (a norma de gerenciamento de riscos de dispositivos médicos). Ela também adicionou requisitos específicos para gerenciamento da cadeia de suprimentos, rastreabilidade de produtos e controle de software. Esta versão se alinhou mais de perto com as tendências regulatórias globais e removeu cláusulas redundantes que se sobrepunham à ISO 9001.
Em 9 de maio de 2025, a ISO lançará oficialmente a ISO 13485:2025, marcando a entrada do sistema de gestão da qualidade para dispositivos médicos em uma nova era de “digitalização, inteligência e conformidade de cadeia completa”. Esta revisão é uma atualização abrangente da edição de 2016 e se concentra em quatro áreas principais:
- A adição de cláusulas sobre ética e segurança de dados para dispositivos médicos baseados em IA, adaptando-se a novas tecnologias como Software como Dispositivo Médico (SaMD) e dispositivos médicos impressos em 3D.
- Fortalecimento do controle de registros eletrônicos, incluindo o uso de blockchain e outras tecnologias para prevenir a adulteração de dados, em conformidade com a FDA 21 CFR Parte 11 e o Anexo 11 da UE.
- Gerenciamento da cadeia de suprimentos aprimorado, implementando auditorias escalonadas e inspeções obrigatórias no local, estendendo o controle aos fornecedores de matérias-primas.
- Extensão do rastreabilidade para 10 anos e aprimoramento dos mecanismos de farmacovigilância (PV).
A estrutura de cláusulas da ISO 13485 foca em ’conformidade regulatória, controle de risco e rastreabilidade“, cobrindo 8 capítulos principais. Ela pode ser dividida em cinco módulos de gestão centrais e quatro requisitos de controle especiais, equilibrando aplicabilidade geral com necessidades específicas da indústria. A versão de 2025 fortalece ainda mais a digitalização e a adaptabilidade a novas tecnologias, tornando-se um foco chave para auditorias.
O requisito central é que as organizações estabeleçam uma estrutura clara de gestão da qualidade, definam papéis e responsabilidades, e garantam que a alta administração esteja comprometida com a conformidade regulatória e a alocação de recursos.
A versão de 2025 adiciona um novo requisito: o CEO deve presidir revisões trimestrais de conformidade regulatória, incluindo avaliações da conformidade com a ética da IA. Também exige que as empresas estabeleçam políticas e metas de qualidade que sejam mensuráveis, acionáveis e diretamente ligadas aos riscos de segurança do produto e aos requisitos regulatórios. Além disso, deve ser estabelecido um mecanismo de revisão gerencial para avaliar regularmente a eficácia do sistema, abordando riscos de conformidade e questões de qualidade prontamente.
Este módulo foca nos cinco recursos principais: pessoas, equipamentos, materiais, métodos e meio ambiente.
Para o pessoal, posições-chave (como inspeção de esterilização, avaliação de risco e revisão de produto) devem ser ocupadas por indivíduos com qualificações reconhecidas pela NMPA (Administração Nacional de Produtos Médicos da China), e treinamento regular sobre regulamentações de dispositivos médicos e habilidades de controle de qualidade deve ser realizado, com registros completos mantidos.
Em relação à infraestrutura, os equipamentos de produção e inspeção devem ser calibrados e mantidos regularmente para garantir a precisão. Equipamentos especializados (como impressão 3D e ferramentas de detecção por IA) devem possuir um processo de verificação digital implementado. Para o ambiente de trabalho, a fabricação de produtos estéreis deve ser realizada em salas limpas certificadas, com a versão de 2025 exigindo monitoramento em tempo real de temperatura e umidade e upload de dados baseado em nuvem para rastreabilidade. Adicionalmente, um mecanismo de garantia de recursos deve estar em vigor para assegurar que os fundos, tecnologia e pessoal necessários estejam disponíveis para controle de qualidade, evitando riscos de qualidade devido a recursos insuficientes.
Este é o módulo central da ISO 13485, cobrindo todo o processo desde o projeto e desenvolvimento até a aquisição, fabricação e serviços pós-venda, com requisitos de conformidade específicos para cada etapa:
- Design e Desenvolvimento: Um processo abrangente de controle de projeto deve ser estabelecido, especificando entradas de projeto (incluindo metas de segurança do paciente), saídas, revisões e requisitos de validação/verificação. A versão de 2025 adiciona requisitos de validação de algoritmos de IA e registros de parâmetros de impressão 3D. A documentação de controle de projeto (DHF) deve incluir dados sobre testes de biocompatibilidade e validação de algoritmos para garantir que os projetos atendam aos padrões regulatórios e de segurança.
- Compras: Os fornecedores devem ser avaliados e controlados por meio de avaliações em níveis, com os fornecedores de alto risco passando por auditorias no local. Os fornecedores devem demonstrar rastreabilidade UDI (Identificação Única de Dispositivo) e assinar acordos de qualidade que definam padrões de qualidade de materiais e requisitos de rastreabilidade. Os implantes devem rastrear materiais até o nível da matéria-prima.
- Produção: Procedimentos Operacionais Padrão (POPs) devem ser desenvolvidos para controlar os parâmetros do processo. A “rastreabilidade de unidade única” (cobertura completa de UDI) deve ser implementada para eliminar desvios durante a produção. Para produtos estéreis, os processos de esterilização devem ser estritamente controlados, com registros completos de esterilização retidos.
- Serviços: Um processo de serviço pós-comercialização deve ser estabelecido para lidar prontamente com reclamações de clientes e questões de qualidade. O feedback deve ser coletado para vigilância pós-comercialização, com a versão de 2025 exigindo que os registros de serviço sejam vinculados ao sistema de rastreabilidade do produto para rastreabilidade completa do ciclo de vida.
As empresas devem estabelecer mecanismos de monitoramento e análise para acompanhar continuamente a eficácia do sistema de gestão da qualidade, a qualidade do produto e a conformidade regulatória:
- As inspeções devem ser realizadas em cada lote de produtos, com os registros de inspeção mantidos por pelo menos cinco anos.
- Um sistema para coleta e comunicação de eventos adversos deve ser estabelecido, com eventos adversos reportados em até 24 horas para as plataformas regulatórias.
- A análise de dados deve ser utilizada para identificar riscos de qualidade e conformidade, com relatórios analíticos servindo como base para ações corretivas.
- Um sistema de ação corretiva e preventiva (CAPA) deve estar implementado para tratar de problemas identificados, com responsabilidades, prazos e requisitos de verificação claros. A versão de 2025 exige que a análise das tendências de eventos adversos seja incluída nas revisões gerenciais para aprimorar as capacidades de alerta de risco.
As empresas devem estabelecer um sistema de gestão de recolhimento de produtos, definindo processos de recolhimento, responsabilidades e planos de emergência. Um exercício simulado de recolhimento deve ser realizado pelo menos uma vez por ano, e as taxas de recolhimento devem ser incluídas nas revisões anuais de gestão. Adicionalmente, as melhorias contínuas do sistema de qualidade devem ser baseadas em revisões de gestão, análise de dados e feedback de clientes, garantindo um ciclo de “conformidade-otimização-reconformidade”. Diferentemente da ISO 9001, o foco aqui é mais na manutenção da conformidade regulatória do que puramente na melhoria de desempenho. O objetivo principal é garantir que o sistema de gestão da qualidade permaneça atualizado com os requisitos regulatórios mais recentes, minimizando os riscos de conformidade.
Desde a versão de 2016, a ISO 13485 exige que as empresas integrem a ISO 14971 (a norma de gerenciamento de riscos de dispositivos médicos) em seus sistemas de gestão da qualidade. A versão de 2025 refina ainda mais os requisitos para o controle de riscos. A lógica fundamental é o “gerenciamento de riscos ao longo do ciclo de vida”, que abrange a identificação, avaliação e controle de riscos desde o projeto até a produção e o monitoramento pós-comercialização. As medidas de controle de riscos devem ser documentadas, verificáveis e rastreáveis.
Para dispositivos baseados em IA, a versão de 2025 introduz o requisito de “classificação de risco de algoritmos”, onde algoritmos de alto risco devem passar por verificação de terceiros. Para dispositivos implantáveis, é exigido monitoramento de risco de longo prazo por até 10 anos, com a manutenção dos dados de risco.
Para dispositivos médicos estéreis (como seringas, implantes articulares, curativos estéreis), a ISO 13485 estabelece requisitos de controle específicos. A versão de 2025 aprimora o controle sobre ambientes limpos e processos de esterilização:
- Salas limpas devem atender aos padrões ISO 14644, e os níveis de limpeza devem ser definidos de acordo com as classificações de risco do produto. O monitoramento em tempo real de partículas suspensas e microrganismos é exigido, com os dados sendo carregados em plataformas regulatórias.
- Os processos de esterilização devem utilizar métodos validados (como vapor ou irradiação), com os parâmetros de esterilização e relatórios de validação retidos para garantir a repetibilidade e rastreabilidade do processo de esterilização.
- Um sistema de barreira estéril deve ser implementado para prevenir a contaminação durante o armazenamento e transporte, e os materiais de embalagem para produtos estéreis devem passar por testes de biocompatibilidade.
A Identificação Única de Dispositivo (UDI) é central para o sistema de rastreabilidade da ISO 13485. A versão de 2025 fortalece a aplicação da UDI ao longo do ciclo de vida do produto:
- Cada dispositivo médico deve receber um código UDI único, incluindo identificadores de produto e produção, em conformidade com bancos de dados UDI globais (como o GUDID dos EUA e o EUDAMED da UE).
- A UDI deve ser exibida na menor embalagem vendável do produto e nas instruções de uso, e para implantes, a UDI deve ser incorporada ao próprio dispositivo.
- Um sistema de rastreabilidade deve ser estabelecido para rastrear o dispositivo desde as matérias-primas, passando pela produção, armazenamento, logística, uso clínico e descarte. Os dados de rastreabilidade devem ser retidos por pelo menos 10 anos para atender aos requisitos regulatórios de rastreabilidade.
Com a aceleração da transformação digital na indústria de dispositivos médicos, a ISO 13485:2025 adiciona diversas cláusulas sobre registros eletrônicos e segurança de dados para se alinhar aos requisitos regulatórios digitais globais:
- Os registros eletrônicos devem ser armazenados em formatos à prova de adulteração (como tecnologia blockchain), com controle de acesso e logs de operação registrados por pelo menos 10 anos, em conformidade com a FDA 21 CFR Parte 11 e o Anexo 11 da UE.
- Para dispositivos médicos de IA e software médico, um sistema de gestão de segurança de dados deve estar em vigor para prevenir violações de dados, proteger a privacidade do paciente e garantir a conformidade com o GDPR e outras leis de proteção de dados.
- Um mecanismo de backup de dados e recuperação de emergência deve ser implementado para garantir que os dados não sejam perdidos e possam ser restaurados em caso de emergência.
A ISO 13485 não se aplica apenas a fabricantes de dispositivos médicos, mas abrange toda a cadeia da indústria de dispositivos médicos. Qualquer organização envolvida no projeto, desenvolvimento, produção, aquisição, vendas, serviços, esterilização, armazenamento ou descarte de dispositivos médicos deve cumprir os requisitos desta norma. As entidades específicas aplicáveis incluem:
Estes incluem fabricantes de dispositivos médicos ativos (como ventiladores, monitores), dispositivos médicos passivos (como seringas, implantes articulares), reagentes de diagnóstico in vitro (como kits de teste de ácido nucleico, tiras de teste de glicose), software médico e dispositivos médicos baseados em IA (como software de telemedicina, equipamentos de diagnóstico por imagem com IA) e dispositivos médicos impressos em 3D. Essas entidades são o grupo central ao qual a norma se aplica e devem atender integralmente a todos os requisitos da norma.
Isso inclui fornecedores de matérias-primas de dispositivos médicos (como plásticos médicos, materiais metálicos), fornecedores de componentes (como sensores, chips), fornecedores de materiais de embalagem, prestadores de serviços de esterilização e empresas de logística. Essas entidades devem atender aos requisitos da norma relacionados à gestão da cadeia de suprimentos, rastreabilidade e controle estéril, e cooperar com os fabricantes principais para completar as auditorias de conformidade.
Isso inclui distribuidores de dispositivos médicos, agentes, organizações de serviços pós-venda, instituições clínicas (como hospitais e clínicas) e organizações de descarte e reciclagem de dispositivos médicos. Tais entidades devem atender aos requisitos de rastreabilidade, serviço pós-venda e notificação de eventos adversos definidos na norma para garantir a conformidade e a segurança na distribuição e uso dos produtos.
A ISO 13485 também se aplica a cenários de fabricação por contrato e pesquisa por contrato, onde a parte contratante e o contratado devem definir claramente suas respectivas responsabilidades de qualidade. O contratado deve possuir um sistema de gestão da qualidade que atenda à norma, e a parte contratante deve auditar e gerenciar o sistema de qualidade do contratado. Adicionalmente, a norma se aplica a empresas de exportação de dispositivos médicos, que devem estabelecer um sistema de qualidade baseado nos requisitos da ISO 13485 para atender aos requisitos regulatórios de entrada do mercado-alvo.
A certificação ISO 13485 é voluntária, mas, por ser considerada um pré-requisito para acesso ao mercado na maioria dos países, muitas empresas buscam proativamente a certificação. O processo de certificação é dividido em seis etapas e deve estar alinhado aos requisitos da norma para garantir a implementação eficaz do sistema:
A empresa deve, primeiramente, definir o escopo da certificação (como categorias de produtos e processos envolvidos), revisar os requisitos regulatórios do mercado-alvo (como NMPA na China, MDR na UE, QSR 820 nos EUA) e estabelecer um plano de construção do sistema de qualidade com base na versão mais recente da ISO 13485 (2025). A empresa também deve formar uma equipe especializada, definir papéis e responsabilidades, e conduzir treinamentos sobre a norma e as regulamentações para garantir que todo o pessoal relevante compreenda os requisitos centrais da norma.
A empresa deve compilar os documentos do sistema de qualidade com base na norma e na situação real da empresa. Esses documentos geralmente incluem o manual da qualidade, procedimentos, instruções de trabalho e formulários. Os documentos devem abranger todas as cláusulas da norma e ser adaptados às características dos produtos e aos processos de produção da empresa. Após a conclusão da documentação, o sistema de qualidade deve ser implementado, com um ciclo operacional mínimo de 3 meses. Durante este período, devem ser mantidos registros completos de operação (como registros de produção, registros de inspeção, registros de avaliação de risco e registros de revisão gerencial) para verificar a viabilidade e a eficácia do sistema.
A empresa deverá formar uma equipe de auditoria interna e realizar auditorias internas com base nos requisitos da ISO 13485. O foco da auditoria é avaliar a conformidade dos documentos do sistema, a eficácia da implementação do sistema e identificar quaisquer lacunas de conformidade ou problemas de qualidade. Após a identificação de problemas, deverão ser desenvolvidas ações corretivas e preventivas. Uma vez implementadas as ações corretivas, os relatórios de auditoria e os registros das ações corretivas deverão ser retidos para garantir que o sistema atenda aos requisitos da auditoria de certificação.
A alta direção da empresa deve realizar uma análise crítica da gestão, avaliando a eficácia, a adequação e a suficiência do sistema de qualidade com base nos resultados de auditorias internas, dados operacionais do sistema, feedback de clientes, atualizações regulatórias e resultados de avaliação de riscos. A empresa deve desenvolver um plano de melhoria do sistema para abordar as deficiências identificadas, e os relatórios da análise crítica da gestão devem ser retidos para garantir que o sistema esteja sempre alinhado com o desenvolvimento da empresa e com os requisitos regulatórios.
A empresa seleciona um organismo de certificação qualificado (como TÜV, SGS ou CQC na China) e submete um pedido de certificação. O pedido deve incluir documentos do sistema de qualidade, registros de operação do sistema, relatórios de auditoria interna, relatórios de análise crítica pela gerência e materiais relacionados ao produto (como certificados de registro do produto, relatórios de inspeção). O organismo de certificação analisará os materiais do pedido e, se aprovado, agendará uma auditoria no local.
Auditores do organismo de certificação visitarão as instalações da empresa para verificar a implementação efetiva do sistema de qualidade, com foco em áreas como o setor de produção, laboratório de inspeção e almoxarifado. Os auditores revisarão os registros operacionais e entrevistarão o pessoal relevante para determinar se o sistema está em conformidade com a ISO 13485. Caso não conformidades sejam encontradas durante a auditoria no local, a empresa deverá corrigi-las dentro de um prazo especificado e apresentar um relatório de correção com materiais de verificação. Assim que as ações corretivas forem verificadas como eficazes, o organismo de certificação emitirá a certificação ISO 13485. O certificado terá validade de 3 anos, período durante o qual auditorias de acompanhamento anuais são exigidas. Após 3 anos, um processo de recertificação deverá ser realizado.
Praticantes frequentemente confundem ISO 13485, ISO 9001 e GMP, pois todas as três estão relacionadas ao controle de qualidade. No entanto, existem diferenças significativas em seu posicionamento, escopo e requisitos centrais, bem como algumas conexões entre elas. É essencial distingui-las claramente:
Conexão:
Ambas as normas ISO 13485 e ISO 9001 são baseadas no ciclo PDCA (Planejar-Fazer-Verificar-Agir), visando a melhoria da gestão da qualidade através de processos padronizados. A ISO 13485 empresta sua estrutura fundamental da ISO 9001, e as empresas que obtiveram a certificação ISO 13485 podem reutilizar alguns de seus documentos de sistema para reduzir a carga de trabalho da certificação ISO 9001.
Diferenças:
- Posicionamento: A ISO 9001 é uma norma geral de gestão da qualidade aplicável a todas as indústrias, com foco principal em “satisfação do cliente e melhoria contínua”. A ISO 13485 é uma norma especializada para dispositivos médicos, com foco em “conformidade regulatória e controle de riscos”.”
- Diferenças de Cláusula: A ISO 13485 elimina cláusulas não relacionadas à indústria de dispositivos médicos que estão presentes na ISO 9001 e adiciona cláusulas especializadas para gerenciamento de risco, rastreabilidade UDI, controle estéril, etc., com requisitos mais rigorosos para conformidade regulamentar.
- Cenários Aplicáveis: A ISO 9001 pode ser utilizada como uma ferramenta geral para a melhoria da gestão da qualidade, enquanto a ISO 13485 é um pré-requisito crítico para o acesso ao mercado de empresas de dispositivos médicos.
Conexão:
As Boas Práticas de Fabricação (BPF) na indústria farmacêutica possuem um correspondente na indústria de dispositivos médicos – BPF para dispositivos médicos. Ambos os padrões visam garantir a “qualidade e segurança do produto”, enfatizando o controle de processos, o controle estéril e a rastreabilidade na produção. A certificação ISO 13485 pode servir como uma referência importante para inspeções de BPF, e muitas empresas estabelecerão um sistema de qualidade que atenda aos requisitos de ambos os padrões simultaneamente.
Diferenças:
- Natureza As BPF (Boas Práticas de Fabricação) são um requisito obrigatório, e os fabricantes de dispositivos médicos devem cumprir os padrões de BPF para obter uma licença de produção. A ISO 13485 é uma certificação voluntária, embora seja um pré-requisito para acesso ao mercado em muitos países.
- Escopo: GMP foca no controle de qualidade durante a produção, enquanto a ISO 13485 abrange todo o ciclo de vida, incluindo projeto, desenvolvimento, aquisição, vendas e serviços.
- Foco de Controle: O GMP foca mais na conformidade do processo de produção, com controles detalhados sobre o ambiente de produção, equipamentos e pessoal. A ISO 13485, por outro lado, enfatiza a integridade geral do sistema, com foco no controle de risco e na colaboração regulatória em toda a cadeia.
Em muitos países e regiões, a certificação ISO 13485 é um pré-requisito para a entrada no mercado de dispositivos médicos. Por exemplo, o MDR da UE exige que os fabricantes de dispositivos médicos implementem um sistema de qualidade compatível com a ISO 13485 para obterem a certificação CE. A FDA reconhece a ISO 13485 como um padrão equivalente ao QSR 820, simplificando o processo de registro da FDA. Na China, a NMPA não exige a certificação ISO 13485, mas é um fator chave nas verificações de registro de dispositivos médicos e de licença de produção. Obter a certificação aumenta a probabilidade de aprovação nessas verificações. Além disso, a ISO 13485 é um padrão reconhecido globalmente, e a certificação reduz os custos de conformidade para empresas que entram em mercados internacionais.
A ISO 13485 foca no gerenciamento de riscos do ciclo de vida, auxiliando as empresas a identificar, avaliar e controlar riscos de qualidade e conformidade em todo o ciclo de vida do produto. Ela reduz desvios e produtos não conformes durante a produção, minimiza custos de retrabalho e sucata, e mitiga o risco de multas, recalls de produtos e disputas legais decorrentes de não conformidade regulatória, especialmente para produtos de alto risco como dispositivos médicos de IA e implantes.
A certificação ISO 13485 comprova o compromisso da empresa com a qualidade e a conformidade regulatória, aumentando sua credibilidade no mercado de dispositivos médicos. Melhora o reconhecimento da marca da empresa, atrai clientes de alta qualidade e aumenta a confiança nos produtos da empresa. Particularmente para empresas globais, a ISO 13485 é frequentemente considerada um “passaporte de negócios global”, aumentando as oportunidades comerciais.
Através da implementação da ISO 13485, as empresas otimizam seus processos internos, aprimoram a eficiência, reduzem desperdícios e elevam a qualidade dos produtos. O estabelecimento de um sistema de gestão da qualidade orientado à conformidade permite melhoria contínua e mecanismos de resolução de problemas para auxiliar as empresas a otimizar os processos de projeto, desenvolvimento, produção e serviço.
Muitas empresas enfrentam inúmeros desafios durante o processo de estabelecimento e certificação da ISO 13485, especialmente com os novos requisitos da versão de 2025. É essencial desenvolver estratégias direcionadas para garantir a implementação eficaz do sistema:
Os requisitos regulatórios para dispositivos médicos diferem significativamente entre as regiões, incluindo o MDR da UE, o QSR 820 da FDA dos EUA e os regulamentos NMPA da China. As empresas enfrentam desafios para construir um sistema de qualidade que atenda aos requisitos regulatórios globais, especialmente com as novas cláusulas relacionadas à digitalização e IA na versão de 2025, que exigem alinhamento com múltiplos quadros regulatórios nacionais.
Estratégia:
Forme uma equipe regulatória profissional para identificar as diferenças entre as regulamentações nos mercados-alvo. Desenvolva soluções de conformidade diferenciadas que integrem os requisitos da norma ISO 13485:2025. Colabore com empresas de consultoria terceirizadas para avaliações de adaptação regulatória e atualizações pontuais dos documentos do sistema de qualidade. Estabeleça um mecanismo de alerta de atualizações regulatórias para rastrear as mudanças regulatórias globais e otimizar o sistema de qualidade de acordo.
Com o rápido desenvolvimento da IA e de dispositivos médicos impressos em 3D, as empresas lutam para manter um controle de qualidade adequado sobre essas novas tecnologias, falhando em atender aos requisitos adicionais da versão de 2025, como validação de algoritmos, registro de parâmetros e segurança de dados.
Estratégia:
Introduzir equipes técnicas especializadas para estabelecer processos de controle para novas tecnologias, como procedimentos de validação de algoritmos de IA e processos de controle de parâmetros de impressão 3D. Trabalhar com organizações terceirizadas para realizar validação de algoritmos e testes de biocompatibilidade para garantir a conformidade. Fortalecer o treinamento do pessoal técnico para aprimorar sua capacidade de controlar a qualidade de novas tecnologias e adaptar-se aos novos requisitos da norma.
Algumas empresas focam apenas em sua própria conformidade, negligenciando, assim, o controle das partes a montante e a jusante da cadeia de suprimentos. Auditorias insuficientes de qualificação de fornecedores e dados incompletos de rastreabilidade tornam impossível a rastreabilidade de cadeia completa, especialmente com os requisitos mais rigorosos de auditoria da cadeia de suprimentos introduzidos na versão de 2025.
Estratégia:
Estabelecer um sistema de gerenciamento de fornecedores em níveis para conduzir auditorias diferenciadas com base no nível de risco dos fornecedores, com fornecedores de alto risco exigindo auditorias no local. Assinar acordos de qualidade com os fornecedores para esclarecer os requisitos de rastreabilidade e conformidade, e exigir que os fornecedores forneçam a certificação ISO 13485 ou provas de conformidade. Construir uma plataforma de rastreabilidade da cadeia de suprimentos para permitir a comunicação de dados com os fornecedores, garantindo a rastreabilidade de toda a cadeia de matérias-primas e componentes.
Algumas empresas criam documentos de sistema superficialmente em conformidade apenas para obter a certificação, sem integrar os requisitos do sistema às operações de produção reais. Como resultado, o sistema torna-se uma formalidade e falha em gerenciar riscos de forma eficaz, muitas vezes levando a não conformidades durante as auditorias.
Estratégia:
Construa o sistema para que esteja alinhado com os processos de produção reais, a fim de evitar implementações “baseadas em modelos”. Forneça treinamento abrangente para todo o corpo de funcionários, assegurando que cada posição compreenda os requisitos específicos do sistema para a sua função. Estabeleça um mecanismo regular de auditoria interna do sistema para revisar periodicamente a operação do sistema, identificar quaisquer discrepâncias com as práticas reais e corrigi-las de forma atempada para melhorar a eficácia do sistema.
A ISO 13485, como norma de sistema de gestão da qualidade para a indústria de dispositivos médicos, seu valor central reside não em meramente “obter um certificado”, mas em alcançar a conformidade regulatória, o controle de riscos e a estabilidade da qualidade por meio de um sistema de gestão padronizado em toda a cadeia, assegurando a saúde e a segurança dos pacientes. Da estrutura básica da versão de 1996 às atualizações digitais e inteligentes da versão de 2025, a ISO 13485 sempre acompanhou os desenvolvimentos da indústria, tornando-se um suporte central para as operações conformes e a competição de mercado das empresas globais de dispositivos médicos.
Na PCBCool, compreendemos a importância crucial de cumprir a ISO 13485 para fabricantes de dispositivos médicos. Fazer parceria com um fabricante certificado ISO 13485 confiável como nós para o seu Soluções em eletrônica médica garante não apenas a conformidade regulatória, mas também aprimora a qualidade e a confiabilidade de seus produtos. Desde a fase de projeto até a fabricação, montagem e integração do produto final, a PCBCool oferece soluções abrangentes de ponta a ponta. Nossa vasta experiência e compromisso com a precisão nos permitem entregar dispositivos médicos de alta qualidade e em conformidade, que atendem aos padrões em evolução do setor. Esteja você desenvolvendo novos dispositivos ou escalando a produção, nossa expertise o apoiará em cada etapa do processo.